
En un mot
Les principaux développements de cette année concernent l’amélioration de l’accès aux traitements, la clarification de l’étiquetage et la mise à jour des recommandations cliniques. L’eskétamine a obtenu l’autorisation de mise sur le marché en monothérapie pour la dépression résistante au traitement. La lumatepérone est devenue une option thérapeutique adjuvante pour le trouble dépressif majeur. Les nouvelles recommandations de l’APA déconseillent l’utilisation d’antipsychotiques pour la prévention du délire. Enfin, les premières recommandations conjointes sur la réduction progressive des benzodiazépines insistent sur l’importance de réductions hyperboliques.

Ce qui a changé en 2025 (Mises à jour à haut rendement)
- Délire: Les nouvelles recommandations de l’APA déconseillent les antipsychotiques en prévention ou en traitement de routine ; il est préférable de les réserver aux situations de détresse sévère ou de comportements à risque, après avoir épuisé les stratégies non pharmacologiques et pris en compte les facteurs réversibles.
- Benzodiazépines: Les recommandations conjointes concernant la réduction progressive des doses insistent sur une diminution individualisée et graduelle ; il convient d’éviter un arrêt brutal et d’envisager des réductions « hyperboliques » à mesure que les doses diminuent.
- Dépression résistante au traitement (DRT) : L’eskétamine (Spravato) a obtenu l’autorisation de mise sur le marché en monothérapie pour la DRT ; il s’agit principalement d’une modification visant à assouplir l’indication plutôt que d’une nouvelle allégation d’« efficacité supérieure » (les programmes de gestion des risques, l’organisation du travail en clinique et les obstacles à l’accès aux soins continuent d’influencer l’utilisation en pratique clinique).
- Trouble dépressif majeur (TDM) : La lumatépérone (Caplyta) a rejoint les options de traitement adjuvant approuvées par la FDA pour le TDM, ajoutant ainsi une nouvelle alternative antipsychotique atypique aux traitements adjuvants déjà existants.
- Maintien du traitement du trouble bipolaire: La suspension injectable à libération prolongée de rispéridone pour administration sous-cutanée (UZEDY) a été approuvée en monothérapie ou en traitement adjuvant au lithium ou au valproate pour le traitement d’entretien du trouble bipolaire I chez l’adulte.
- Pour le traitement d’entretien du trouble bipolaire, utilisez le schéma mensuel (le schéma bimestriel n’est pas recommandé selon la notice).
- Schizophrénie: Une réduction historique de la charge administrative..
- Les pharmacies n’ont plus besoin d’être inscrites au programme REMS pour délivrer des médicaments ; les prescripteurs n’ont pas besoin d’enregistrer les patients dans une base de données fédérale.
- Les prescripteurs doivent continuer de surveiller la numération absolue des neutrophiles (ANC) en lien avec l’avertissement encadré concernant la neutropénie.
- Dépendance: La mise à jour de l’étiquetage concernant l’initiation rapide de la buprénorphine à libération prolongée (Sublocade) vise à remédier à un point de décrochage observé en pratique clinique, en permettant la transition vers l’injection après une exposition transmucosale minimale, plutôt que d’exiger une phase préliminaire prolongée de « tolérance ».
- Fibromyalgie: La cyclobenzaprine sublinguale (Tonmya) a obtenu son approbation sous la forme d’une formulation sublinguale à faible dose ciblant le cycle sommeil-douleur.
- Maladie d’Alzheimer:
- La titration plus lente du donanemab (Kisunla) vise à réduire le risque d’ARIA-E tout en maintenant les effets sur les biomarqueurs modificateurs de la maladie.
- Lumipulse est le premier test sanguin approuvé par la FDA pour faciliter le diagnostic de la maladie d’Alzheimer chez les adultes présentant des troubles cognitifs.
- TDAH: L’étiquetage des stimulants à libération prolongée de la FDA, applicable à l’ensemble de la classe, ajoute un signal de « limitation d’utilisation » plus clair pour les enfants de moins de 6 ans, reflétant une exposition plus élevée et des taux plus élevés d’événements indésirables (y compris une perte de poids cliniquement significative).
Mises à jour 2025 des lignes directrices cliniques
Délire : ligne directrice de pratique de l’APA (septembre 2025)
- Les recommandations de l’APA concernant le délirium constituent la première mise à jour exhaustive depuis plus de 20 ans [1,2].
- Évolution majeure : ces recommandations déconseillent l’utilisation d’antipsychotiques à des fins de prévention du délirium ou pour en accélérer la résolution, réservant ces médicaments aux symptômes neuropsychiatriques sévères, lorsque les seuils de sécurité ou de détresse sont atteints.
- Points clés :[2]
- Usage restrictif des antipsychotiques :
- Les antipsychotiques ne doivent pas être utilisés pour prévenir le délirium ni pour en accélérer la résolution.
- Les antipsychotiques ne doivent être utilisés pour traiter les troubles neuropsychiatriques que si tous les critères suivants sont remplis :
- Les stratégies de désescalade verbale et non verbale ont échoué.
- Les facteurs contributifs (p. ex. douleur, infection) ont été évalués et pris en charge.
- Les symptômes causent une détresse significative au patient ou présentent un risque de préjudice physique pour le patient ou pour autrui.
- Éviter les benzodiazépines et certains « somnifères » :
- Les benzodiazépines ne doivent pas être utilisées en cas de délirium (ni chez les personnes à risque), sauf indication spécifique (par ex. : syndromes de sevrage exclus du champ d’application de la recommandation).
- La mélatonine et le rameltéon ne doivent pas être utilisés pour prévenir ou traiter le délirium.
- Préférence pour la dexmédétomidine en soins intensifs:
- Pour les patients subissant une intervention chirurgicale majeure ou recevant une ventilation mécanique en milieu de soins intensifs, l’APA suggère d’utiliser la dexmédétomidine plutôt que d’autres agents sédatifs pour prévenir le délirium.
- Usage restrictif des antipsychotiques :
Sevrage progressif des benzodiazépines : Recommandations de pratique clinique conjointes
- Recommandation de pratique clinique conjointe, élaborée sur la base d’une revue systématique et d’un consensus clinique par un Comité de recommandations cliniques (CGC) de l’American Society of Addiction Medicine, et approuvée par plusieurs autres sociétés [3].
- Elle présente, à l’intention des cliniciens exerçant dans divers contextes, des recommandations sur la prise en charge et la réduction progressive de l’utilisation des benzodiazépines (BZD).
- Points saillants et principes fondamentaux de la réduction progressive[3]
- Dépendance physique vs. trouble lié à l’usage de substances :
- La dépendance physique est une conséquence biologique attendue de l’utilisation régulière de benzodiazépines et se distingue du trouble de l’usage des benzodiazépines (SUD).
- Quand réduire progressivement
- La réduction progressive est indiquée lorsqu’un patient est susceptible de présenter une dépendance physique et que les risques l’emportent désormais sur les bénéfices (par ex. : chutes, troubles cognitifs, mésusage, co-prescription d’opioïdes).
- Éviter tout arrêt brutal chez tout patient susceptible de présenter une dépendance, en raison du risque de syndrome de sevrage et de convulsions.
- Stratégies de réduction progressive et ajustements
- Les réductions initiales de la dose devraient généralement être de 5 à 10 % toutes les 2 à 4 semaines.
- Le rythme de la diminution ne devrait généralement pas dépasser 25 % toutes les 2 semaines.
- Envisagez un sevrage hyperbolique (réductions de dose absolues plus faibles au fil du temps ; par ex. : 10 mg → 9 mg → 8,1 mg) afin de mieux s’ajuster à l’évolution de l’occupation des récepteurs aux doses plus faibles.
- La réduction hyperbolique est une stratégie de réductions de dose non linéaires et séquentielles, ce qui signifie que l’ampleur des réductions diminue au fil du temps.
- Les patients ayant pris des doses plus faibles pendant une période relativement courte (par ex. < 3 mois) pourraient être en mesure de réduire progressivement leur traitement plus rapidement.
- Agents à action prolongée :
- Envisagez de passer à une benzodiazépine à action prolongée (par ex. diazépam, clonazépam) avant d’entamer la réduction progressive, en l’absence de contre-indications (par ex. dysfonction hépatique sévère).
- Prise en charge des troubles psychiatriques concomitants
- Envisagez sérieusement la réduction progressive des benzodiazépines chez les patients souffrant de TSPT, car ces médicaments sont inefficaces contre les symptômes centraux du TSPT et peuvent accroître le risque de dépression et d’agressivité.
- Dépendance physique vs. trouble lié à l’usage de substances :
Dépression et Troubles de l’Humeur
Esketamine (Spravato) – Approbation en monothérapie (21 janvier 2025)
- Le spray nasal Esketamine (Spravato) CIII a reçu l’approbation de la FDA en tant que monothérapie pour les adultes souffrant de Treatment-Resistant Depression (TRD). Cela représente une approbation de supplemental New Drug Application (sNDA).
- Indication : Dépression résistante au traitement (DRT) chez l’adulte, désormais en monothérapie (voir notre Guide cérébral sur l’eskétamine) :
- L’approbation en monothérapie permet aux cliniciens d’instaurer l’eskétamine sans ajouter un nouvel antidépresseur oral quotidien, ce qui est particulièrement pertinent pour les patients présentant :
- Intolérance significative aux antidépresseurs oraux (p. ex. dysfonction sexuelle, prise de poids, effets indésirables gastro-intestinaux).
- Lourdeur de la charge médicamenteuse ou polypharmacie, rendant l’ajout d’un autre agent oral indésirable.
- L’approbation en monothérapie permet aux cliniciens d’instaurer l’eskétamine sans ajouter un nouvel antidépresseur oral quotidien, ce qui est particulièrement pertinent pour les patients présentant :
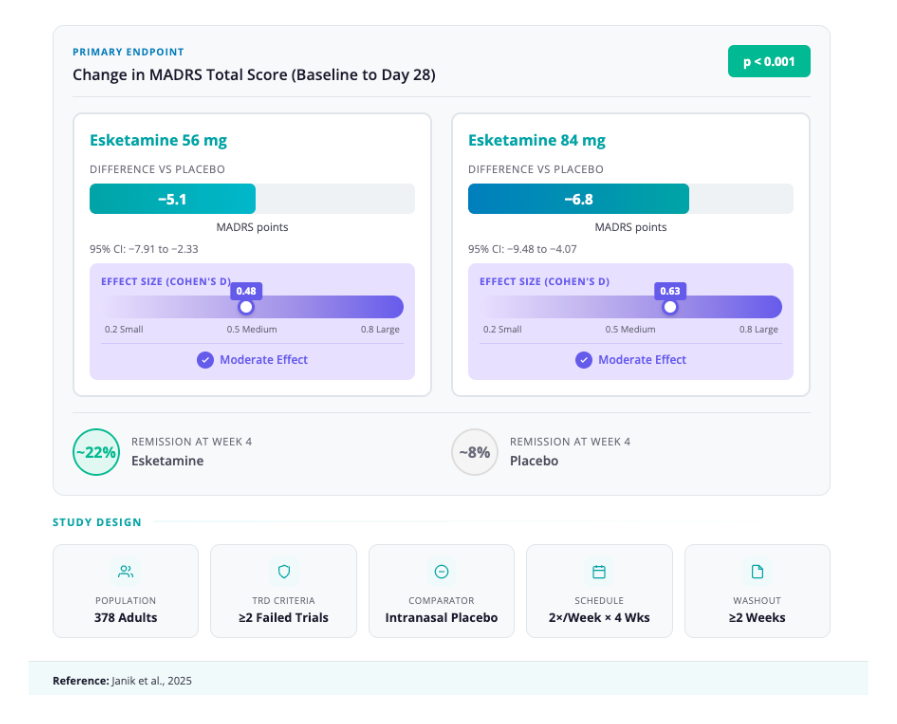
- Essai randomisé pivot post-commercialisation (Étude 3 ; NCT04599855) [4,5]
- Conception : Étude multicentrique, randomisée, en double aveugle et contrôlée par placebo ; 378 adultes souffrant de dépression résistante au traitement (TRD) ont été randomisés selon un ratio 1:1:2 pour recevoir SPRAVATO 56 mg, 84 mg ou un placebo par pulvérisation nasale pendant 4 semaines (deux fois par semaine).
- Critère de jugement principal (MADRS au Jour 28) : Différence des moyennes des moindres carrés par rapport au placebo de −5,1 (56 mg) et −6,8 (84 mg) ; pour les deux doses, P < 0,001.
- Tailles d’effet : 0,48 et 0,63, respectivement.
- Critère d’efficacité secondaire (MADRS au Jour 2, soit environ 24 h) : Les différences intergroupes étaient significatives pour les deux doses d’eskétamine : −3,8 (P = 0,004) pour 56 mg et −3,4 (P = 0,006) pour 84 mg.
- Aspects pratiques :
- Dosing:
- Le schéma thérapeutique demeure inchangé par rapport à l’indication adjuvante (phase d’induction deux fois par semaine, suivie d’une réduction progressive vers une phase d’entretien hebdomadaire ou bimensuelle). [4,5]
- Distribution soumise à des restrictions REMS, incluant une administration en clinique, une surveillance de deux heures et des restrictions de conduite jusqu’au lendemain.
- Les effets indésirables demeurent similaires à ceux observés lors d’une utilisation en traitement d’appoint : dissociation, vertiges, nausées, augmentations transitoires de la pression artérielle et sédation.[5,6]
- Dosing:
- Impact clinique et positionnement :
- Les tailles d’effet observées étaient modestes à modérées (0,48–0,63). Les conclusions concernant la comparaison de ces résultats avec l’eskétamine en association ou d’autres interventions contre la dépression résistante au traitement (DRT) doivent être interprétées avec prudence, car les populations, les méthodologies, les critères d’évaluation et les effets d’attente diffèrent d’un essai à l’autre [7–11].
- Comme pour d’autres interventions à effet psychoactif rapide, la levée de l’aveugle fonctionnel constitue une limite potentielle (les participants et les évaluateurs peuvent déduire l’attribution du traitement à partir des effets subjectifs aigus) ; de manière générale, les comparaisons entre essais doivent être interprétées avec prudence [12].
- En pratique courante, l’administration et la surveillance en clinique, requises par le REMS, ainsi que la planification, le transport et le remboursement demeurent des obstacles majeurs ; ces facteurs influencent souvent les comparaisons en situation réelle avec la kétamine intraveineuse autant que l’efficacité elle-même.
- L’autorisation de la monothérapie élargit principalement la flexibilité d’indication en autorisant l’eskétamine pour la DRT sans association obligatoire avec un antidépresseur oral.
Lumatepérone (Caplyta) – Traitement d’appoint du trouble dépressif majeur (6 novembre 2025)
- La lumatepérone (Caplyta) a reçu l’approbation de la FDA en tant que thérapie adjuvante aux antidépresseurs oraux pour le trouble dépressif majeur (TDM) chez les adultes présentant une réponse inadéquate au traitement antidépresseur standard.[13]
- Indication précédente de la lumatepérone, déjà approuvée par la FDA : [14]
- Schizophrénie (adultes)
- Traitement des épisodes dépressifs associés au trouble bipolaire de type I ou II (dépression bipolaire) chez l’adulte, en monothérapie et en traitement adjuvant au lithium ou au valproate.
- Nouvelle indication : Traitement adjuvant du trouble dépressif majeur (TDM) chez l’adulte.
- D’autres traitements antipsychotiques d’appoint approuvés par la FDA pour le trouble dépressif majeur incluent [15–17]
- Aripiprazole,
- Brexpiprazole,
- Cariprazine,
- Quétiapine à libération prolongée et
- Association olanzapine/fluoxétine
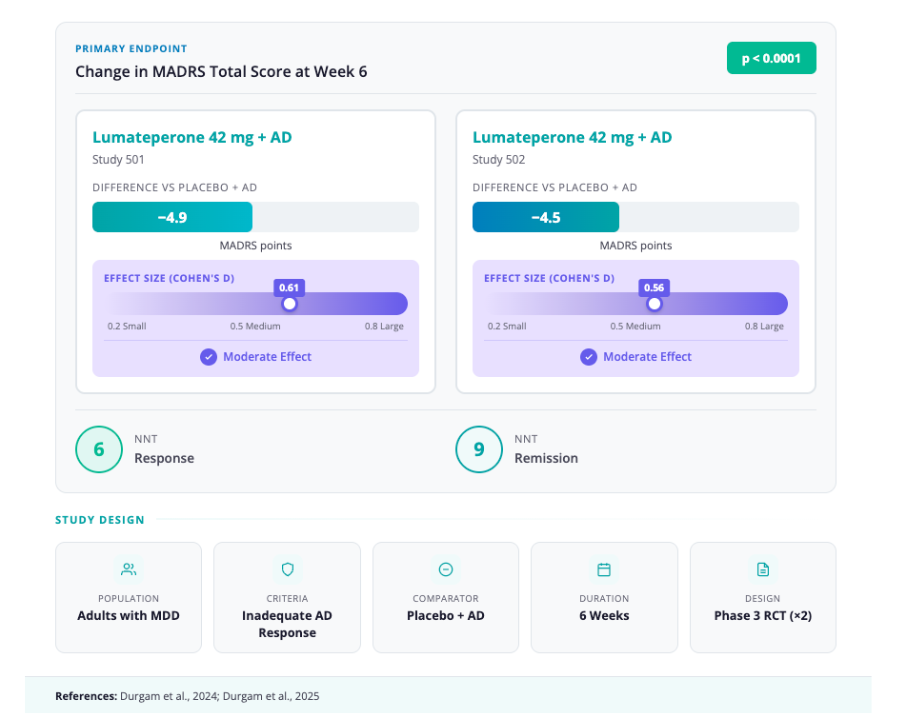
- Données d’efficacité: L’approbation reposait sur deux études de phase 3 (l’étude 501 et l’étude 502). [13,18]
- Conception: Essais mondiaux randomisés, en double aveugle et contrôlés par placebo
- Population: Adultes souffrant de trouble dépressif majeur (TDM) et présentant une réponse inadéquate à un traitement antidépresseur
- Intervention: Lumatepérone 42 mg + Antidépresseur vs Placebo + Antidépresseur
- Durée: 6 semaines
- Critère de jugement principal: Variation du score total MADRS à la 6e semaine
- Résultats clés :
- Étude 501 : réduction de 4,9 points par rapport au placebo (taille de l’effet : 0,61 ; p < 0,0001) [18]
- Étude 502 : réduction de 4,5 points par rapport au placebo (taille de l’effet : 0,56 ; p < 0,0001)
- Nombre de sujets à traiter (NST): environ 6 pour une réponse, 9 pour une rémission. [19]
- Effets secondaires et sécurité :
- Profil métabolique : La lumatepérone présente un profil métabolique favorable. Dans les données de sécurité regroupées, les variations moyennes du poids, de la glycémie et des lipides étaient similaires à celles observées sous placebo [13, 19].
- Effets indésirables fréquents : Vertiges, somnolence, sécheresse buccale, nausées [13, 15, 19].
- Symptômes moteurs : Faible risque de symptômes extrapyramidaux (SEP) ou d’akathisie, comparable à celui du placebo dans les essais cliniques.[13,19]
- Aspects pratiques :
- Posologie : Une dose (42 mg) pour toutes les indications. Aucune titration n’est requise [14].
- Positionnement clinique : En raison de son risque métabolique moindre par rapport à des agents tels que l’olanzapine ou la quétiapine, ce traitement sera probablement privilégié chez les patients présentant des troubles métaboliques préexistants ou ayant pris du poids sous des traitements adjuvants antérieurs [15].
Psychiatrie interventionnelle : TMS chez l’adolescent (août – novembre 2025)
- L’année 2025 a marqué une expansion significative de la stimulation magnétique transcrânienne (SMT) chez les jeunes.
- Suite à l’autorisation accordée en 2024 au système NeuroStar en tant que traitement d’appoint du trouble dépressif majeur (TDM) chez les patients adolescents âgés de 15 à 21 ans [20], la FDA a autorisé, en 2025, plusieurs dispositifs supplémentaires destinés aux adolescents atteints de TDM.
- Approbations:
- MagVenture (MagPro): autorisé le 25 août 2025[21]
- Apollo TMS: Autorisé le 10 septembre 2025 [22]
- BrainsWay Deep TMS (Bobine H): Autorisé le 13 novembre 2025 [23]
- BrainsWay a soumis des données de vie réelle (RWE) portant sur plus de 1 100 patients adolescents, faisant état d’un taux de réponse de 66,1 %. [23]
- Ces approbations ont été largement favorisées par la voie réglementaire 510(k), qui permet l’autorisation de dispositifs en démontrant une « équivalence substantielle » par rapport à un dispositif de référence (en l’occurrence, l’autorisation initiale du NeuroStar) ou par la soumission de données probantes issues de la pratique clinique courante (RWE). [23]
- Protocoles accélérés :
- BrainsWay a également obtenu l’autorisation pour un protocole accéléré destiné aux adultes.
- Protocole standard : 1 séance par jour, 5 jours par semaine pendant 4 semaines (phase aiguë), suivie de 2 séances par semaine pendant une durée pouvant aller jusqu’à 12 semaines (phase de continuation) (Prépublication disponible [24])
- Protocole accéléré : 5 séances par jour pendant 6 jours (phase aiguë), suivies de 2 séances de traitement par jour, un jour par semaine, pendant 4 semaines supplémentaires (phase de continuation) (Prépublication disponible [24])
Stimulation transcrânienne à courant continu (Flow) – Premier dispositif à usage domestique (11 décembre 2025)
- La FDA a autorisé le premier casque de stimulation transcrânienne à courant continu (tDCS) à usage domestique pour le traitement du trouble dépressif majeur (TDM) chez les adultes [25].
- Indication:[25]
- Pour les adultes (18 ans et plus) souffrant de trouble dépressif majeur modéré à sévère.
- Approuvé à la fois en monothérapie et en traitement d’appoint (utilisé en complément d’un traitement médicamenteux).
- Chez les patients non considérés comme réfractaires au traitement médicamenteux.
- Mécanisme:
- Utilise la stimulation transcrânienne à courant continu (tDCS), délivrant un courant électrique faible et constant au cortex préfrontal dorsolatéral (DLPFC) via un casque. [29]
- Données d’efficacité :
- L’approbation s’est appuyée sur un vaste essai randomisé, en double aveugle et contrôlé par placebo (N = 174), récemment publié dans Nature Medicine. [26]
- Résultats:
- Le groupe actif a présenté une réduction significativement plus importante des scores sur l’échelle de dépression de Hamilton (HDRS) par rapport au groupe placebo (p ≈ 0,012 ; taille d’effet de 0,37).
- Les taux de réponse et de rémission ont été supérieurs avec la tDCS active par rapport au placebo, avec un NNT approximatif de 5 pour la réponse et de 4 pour la rémission.
- Le démasquage fonctionnel constitue une préoccupation réelle dans les essais de neuromodulation à domicile ; la FDA signale d’ailleurs explicitement le risque de démasquage — ainsi que la nature mitigée de la littérature scientifique — lors de l’évaluation du rapport bénéfice-risque.
- Aspects pratiques:
- Ce dispositif nécessite une ordonnance aux États-Unis. [25]
- Fourchette de prix : 500 $ – 800 $ (estimation aux États-Unis lors de son lancement au second semestre 2026).
- En Europe, il est déjà disponible au prix de 459 €.
- Protocole: Les patients s’administrent généralement eux-mêmes cinq séances de 30 minutes par semaine au cours des trois premières semaines, suivies d’un programme d’entretien de trois séances par semaine [26].
- Effets secondaires: Généralement légers, incluant des rougeurs ou une irritation cutanée au niveau des électrodes, ainsi que des maux de tête transitoires. Aucun événement indésirable grave n’a été signalé au cours des essais cliniques.[25]
Rispéridone ER (Uzedy) – Trouble bipolaire I (10 octobre 2025)
- La FDA a élargi les indications d’Uzedy — une formulation injectable sous-cutanée à action prolongée (LAI) de rispéridone — afin d’y inclure le traitement d’entretien du trouble bipolaire de type I [30].
- La suspension injectable à libération prolongée de rispéridone pour administration sous-cutanée est indiquée, à raison d’une injection tous les un ou deux mois, pour le traitement de la schizophrénie chez l’adulte. [31]
- Nouvelle indication :
- Monothérapie ou traitement adjuvant au lithium ou au valproate pour le traitement d’entretien du trouble bipolaire I chez l’adulte. [30]
- Conçu pour remédier aux taux élevés de non-observance dans le trouble bipolaire.
- L’absence de phase d’introduction orale peut constituer un avantage logistique dans les contextes de stabilisation aiguë, où l’observance orale ne peut être garantie.
- Injection sous-cutanée :
- Administré par voie sous-cutanée (abdomen ou bras), à l’aide d’une aiguille plus fine que celle utilisée pour les injections intramusculaires traditionnelles dans le fessier ou le deltoïde [30]
- Repose sur une technologie à base de copolymères permettant une libération contrôlée et régulière de la rispéridone [31]
- Aucune phase d’instauration par voie orale : les concentrations plasmatiques thérapeutiques sont atteintes dans les 6 à 24 heures suivant la première injection, éliminant ainsi la nécessité d’une supplémentation orale lors de l’instauration du traitement [31]]
- Aspects pratiques :
- Intervalles d’administration: Disponible selon des options d’administration mensuelle (tous les mois) ou bimestrielle (tous les 2 mois) (50 mg, 75 mg, 100 mg, 125 mg) [31]
- Pour le traitement d’entretien du trouble bipolaire, utiliser le schéma mensuel (le schéma bimestriel n’est pas recommandé selon la notice).[32]
Schizophrénie et psychose
Suppression du REMS de la clozapine (24 février 2025)
- La FDA a supprimé le programme de Stratégie d’évaluation et d’atténuation des risques (REMS) relatif à la clozapine, une décision historique visant à améliorer l’accès au « traitement de référence » pour la schizophrénie résistante au traitement [33].
- Bien que le risque de neutropénie sévère associé à la clozapine persiste, la FDA a établi que le programme REMS pour ce médicament n’est plus nécessaire pour garantir que les bénéfices du traitement l’emportent sur ce risque. [34]
- Auparavant, ce programme imposait aux pharmacies de vérifier l’inscription du patient ainsi que son nombre absolu de neutrophiles (NAN) au sein d’une base de données centralisée avant toute délivrance, créant ainsi, bien souvent, des obstacles administratifs à l’accès aux soins.
- Nouveau flux de travail:
- Prescriptrices(feminine):
- Il reste nécessaire de surveiller la numération absolue des neutrophiles (ANC) conformément à l’étiquetage du produit (par ex. chaque semaine au cours des 6 premiers mois).
- Toutefois, il n’est plus exigé d’inscrire les patients sur le portail REMS ni de signaler chaque valeur d’ANC à la base de données de la FDA pour générer une « autorisation de délivrance ».
- Pharmacies:
- Il n’est pas nécessaire d’être inscrit au programme REMS pour commander de la clozapine auprès de distributeurs grossistes.
- Les pharmaciens ne sont pas tenus de vérifier l’éligibilité des patients — y compris le suivi de la numération absolue des neutrophiles (ANC) — avant de leur délivrer de la clozapine.
- Prescriptrices(feminine):
- Considérations de sécurité continues :
- L’avertissement encadré relatif à la neutropénie sévère (agranulocytose) demeure en vigueur [35].
- La responsabilité clinique du suivi de la numération absolue des neutrophiles (NAN) incombe désormais entièrement aux prescripteurs et à leurs équipes soignantes, par le biais de protocoles internes, plutôt que de reposer sur une vérification par registre imposée au niveau fédéral.
- Cela élimine le « goulot d’étranglement » administratif qui dissuadait souvent les cliniciens de prescrire la clozapine.
Trouble du déficit de l’attention avec hyperactivité (TDAH)
Mise à jour de l’étiquetage pour l’ensemble de la classe : stimulants à libération prolongée (30 juin 2025)
- La FDA a publié une communication sur la sécurité des médicaments exigeant la mise à jour des avertissements concernant tous les produits à libération prolongée (LP) de méthylphénidate et d’amphétamine [36].
- Les étiquettes mises à jour doivent inclure une « Limitation d’utilisation » indiquant que les enfants de moins de 6 ans présentent une exposition plasmatique plus élevée et un taux plus important d’effets indésirables, notamment une perte de poids cliniquement significative.[36]
- Les résultats étant cohérents pour les produits à libération prolongée d’amphétamine et de méthylphénidate, la FDA a conclu que le rapport bénéfice-risque ne justifie pas l’utilisation de formulations à libération prolongée chez les enfants de moins de <6.
- Raisonnement:
- Bien que de nombreux stimulants à libération prolongée ne soient pas approuvés par la FDA pour les enfants de moins de 6 ans, la prescription hors AMM dans cette tranche d’âge a augmenté.[36]
- Implications cliniques pour les prescripteurs:
- Évitez, dans la mesure du possible, l’utilisation de stimulants à libération prolongée (LP) chez les enfants d’âge préscolaire. Pour les enfants de moins de 6 ans actuellement sous une formulation à libération prolongée, réévaluez le rapport bénéfice-risque ; en cas de perte de poids ou de survenue d’autres effets indésirables, la FDA recommande d’interrompre le produit à libération prolongée et/ou de passer à des alternatives (par ex. : stimulants à libération immédiate, thérapie comportementale) [36].
- Pour tout enfant sous traitement stimulant — et plus particulièrement pour ceux de moins de 6 ans —, surveillez régulièrement la taille, le poids et les percentiles de l’IMC, et intervenez précocement en cas de perte de poids ou de ralentissement de la croissance.
Maladie d’Alzheimer
Donanemab (Kisunla) – Calendrier de titration modifié (juillet 2025)
- La FDA a approuvé une mise à jour de l’étiquetage de Kisunla (donanemab-azbt) recommandant un nouveau schéma de titration plus progressif. [37]
- Spécifiquement destiné à réduire le risque d’anomalies d’imagerie liées aux amyloïdes avec œdème/épanchement (ARIA-E) chez les patients atteints de la maladie d’Alzheimer symptomatique au stade précoce.
- L’ARIA peut être détectée par imagerie par résonance magnétique (IRM) et peut se présenter sous la forme d’une ARIA avec œdème (ARIA-E) ou d’une ARIA avec dépôts d’hémosidérine (ARIA-H).[38]
- L’ARIA est généralement asymptomatique, bien que des événements graves et potentiellement mortels puissent survenir.
- Pharmacologie:
- Le donanemab est un anticorps monoclonal anti-amyloïde β administré par voie intraveineuse qui se fixe aux plaques amyloïdes déposées et favorise leur élimination, ralentissant ainsi le déclin cognitif et fonctionnel chez les patients présentant des troubles cognitifs légers ou une démence légère due à la maladie d’Alzheimer. [39]
- Protocole de titration
- Ancien schéma thérapeutique standard : 700/700/700/1400 mg
- Nouveau schéma de titration : Cette mise à jour remplace l’escalade rapide standard par un protocole d’augmentation plus progressif, afin d’atténuer les réponses inflammatoires au cours de la phase initiale du traitement. [38]
- Aspects pratiques et pertinence psychiatrique :
- Le Kisunla est administré par perfusion intraveineuse de 30 minutes toutes les 4 semaines, selon le nouveau schéma de titration et sous surveillance par IRM cérébrales sériées afin de dépister d’éventuels ARIA.
- Stratégie « Treat-to-clear » (traitement jusqu’à élimination) issue de l’étude TRAILBLAZER-ALZ 2 : les cliniciens peuvent interrompre le traitement une fois que les plaques amyloïdes ont été réduites à un niveau minimal, tel que confirmé par TEP, plutôt que de le poursuivre indéfiniment.[40–43]
Lumipulse Plasma (Fujirebio) – Premier test sanguin approuvé par la FDA (16 mai 2025)
- La FDA a autorisé la mise sur le marché du premier dispositif de diagnostic in vitro analysant le sang pour faciliter le diagnostic de la maladie d’Alzheimer[44]
- Indication:
- À utiliser chez les patients adultes âgés de 55 ans et plus présentant des troubles cognitifs et faisant l’objet d’une évaluation pour la maladie d’Alzheimer.
- Mécanisme:
- Le test mesure le rapport entre la p-Tau217 (tau phosphorylée) et la β-amyloïde 1-42 dans le plasma sanguin [LumipulseFDA].
- Ce rapport est corrélé à la présence ou à l’absence de plaques amyloïdes dans le cerveau du patient.
- Données de précision :
- Dans des études de validation clinique portant sur 499 patients, le test a démontré une concordance élevée avec les « étalons de référence » (TEP ou analyse du LCR) [LumipulseFDA].
- Aspects pratiques :
- Aspects pratiques : Accessibilité : Il offre une alternative moins invasive, plus rapide et considérablement moins coûteuse que l’imagerie TEP ou la ponction lombaire.
- Toutefois, le ratio plasmatique Lumipulse G pTau217/ß-Amyloïde 1-42 n’est pas destiné à servir de test de dépistage ou de test diagnostique autonome ; d’autres évaluations cliniques ou des examens complémentaires doivent être utilisés pour déterminer les options thérapeutiques [44].
Dépendance et gestion de la douleur
Cyclobenzaprine sublinguale (Tonmya) – Fibromyalgie (15 août 2025)
- Tonmya (cyclobenzaprine HCl sublingual tablets) received FDA approval for the management of fibromyalgia [45]
- Indication: Management of fibromyalgia in adults
- Pharmacology & Mechanism:
- Tonmya is a sublingual, transmucosal formulation of cyclobenzaprine (2.8 mg or 5.6 mg) designed for rapid absorption at bedtime.
- The sublingual route bypasses first-pass hepatic metabolism, leading to higher parent cyclobenzaprine and lower norcyclobenzaprine exposure than oral tablets [46]
- Norcyclobenzaprine is the active metabolite of cyclobenzapirine, with a half-life of several days (approximately 72hours)
- This is intended to reduce next-day grogginess and prolonged sedation
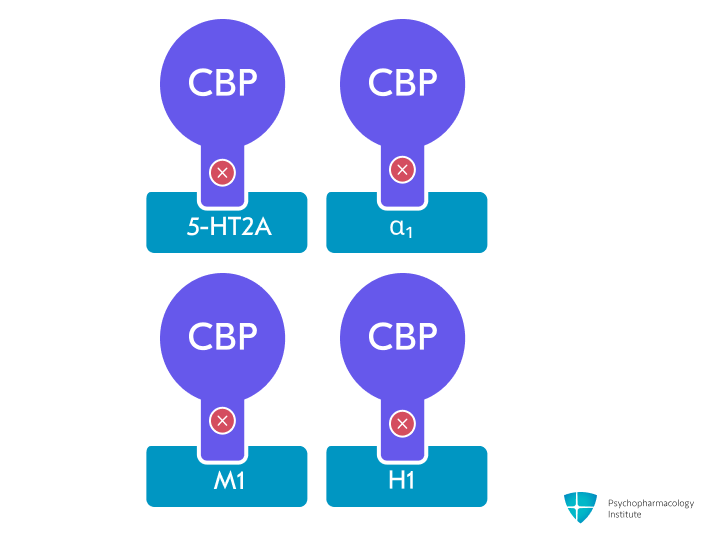
- Cyclobenzaprine antagonizes 5-HT₂A, α₁-adrenergic, M₁-muscarinic, and H₁-histaminergic receptors, modulating sleep architecture and decreasing nonrestorative sleep, which is strongly linked to central sensitization and pain amplification in fibromyalgia [46]
- Norcyclobenzaprine antagonizes the same receptors as cyclobenzaprine but with lower potency
- Norcyclobenzaprine is also a more potent inhibitor of norepinephrine transporters than cyclobenzaprine
- Conceptually, Tonmya is positioned as a centrally acting, sleep-targeted analgesic that may interrupt the cycle of poor sleep → heightened pain → further sleep disruption [46]
- Efficacy data:
- Effect size for pain reduction was ~0.38, similar to duloxetine, pregabalin, and milnacipran (with effect sizes of 0.36, 0.31, and 0.22, respectively) [46–49]
- Safety & Tolerability:
- Overall tolerability was favorable; most adverse events were mild, transient oral effects related to sublingual administration (oral hypoesthesia, paresthesia, altered taste) [46,50]
- Rates of serious adverse events and discontinuations due to AEs were low and comparable to placebo in the Phase 3 program.
- Practical Aspects & Relevance:
- Dosing: Taken once nightly at bedtime, typically after a short titration period (2.8 mg for 2 weeks, then 5.6 mg)
- While cyclobenzaprine is not a novel molecule, FDA approval of this low-dose sublingual formulation will enable broader clinical use. Real-world evidence from post-marketing experience will clarify where it fits within the fibromyalgia treatment landscape [51]
Sublocade (Buprenorphine Extended-Release) – Rapid Initiation (February 24, 2025)
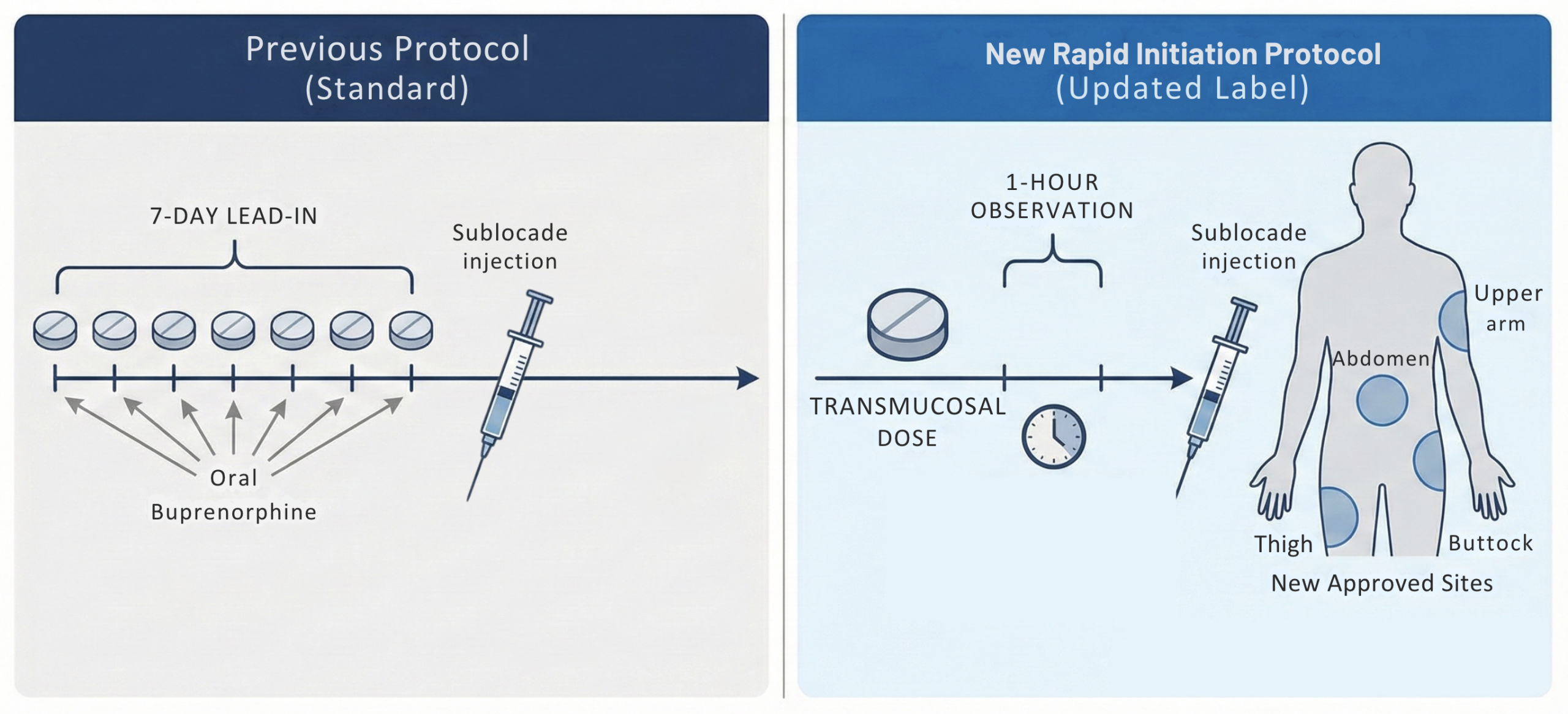
- FDA approved a label update for buprenorphine extended-release injection (Sublocade) that introduces a rapid initiation protocol and alternative injection sites for the treatment of moderate-to-severe opioid use disorder (OUD) [52]
- The previous label required a 7-day “lead-in” period with oral transmucosal buprenorphine to confirm tolerability/clinical appropriateness. This week-long gap often resulted in patient dropout, particularly in emergency department (ED) or crisis settings [53]
- New label: Allows for the administration of the Sublocade injection after just a single dose of transmucosal buprenorphine, followed by a 1-hour observation period prior to injection [52]
- New Injection Sites: The approval also expands administration sites to include the thigh, buttock, and upper arm (previously restricted to the abdomen) [52]
- This added flexibility can be helpful in patients with central obesity, scarring, or prior injection-site issues, and may facilitate administration across diverse clinical settings.
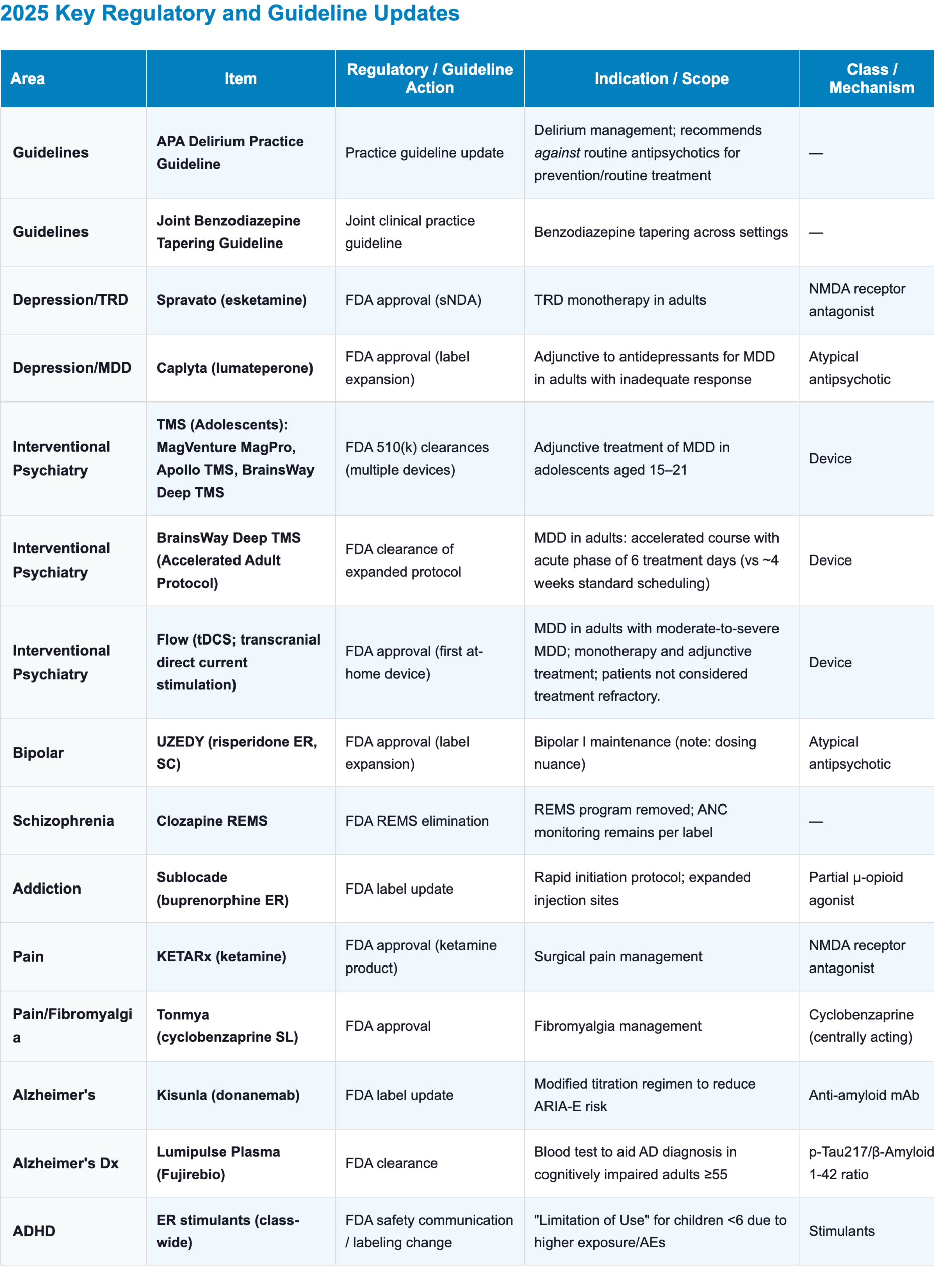
Appendix: 2025 Key Regulatory and Guideline Updates
References
1. The american psychiatric association practice guideline for the prevention and treatment of delirium (Second Edition). (2025). American Psychiatric Publishing. https://doi.org/10.1176/appi.books.9780890428023
2. Crone, C., Fochtmann, L. J., Ahmed, I., Balas, M. C., Boland, R., Escobar, J. I., Heinrich, T., Jackson-Triche, M., Levenson, J. L., Mattison, M., McCullen Truett, J., Oldham, M. A., Seritan, A., Fochtmann, L. J., & Hong, S.-H. (2025). The American Psychiatric Association Practice Guideline for the Prevention and Treatment of Delirium. American Journal of Psychiatry, 182(9), 880–884. https://doi.org/10.1176/appi.ajp.25182013
3. CGC, & ASAM. (2025). Joint CPG on BZD Tapering: Considerations when Benzodiazepine Risks Outweigh Benefits. https://downloads.asam.org/sitefinity-production-blobs/docs/default-source/guidelines/benzodiazepine-tapering-2025/bzd-tapering-document—final-approved-version-for-distribution-02-28-25.pdf?sfvrsn=5bdf9c81_6
4. US Food and Drug Administration (2025). SPRAVATOreg esketamine hydrochloride solution Janssen Pharmaceuticals Inc. Prescribing Information.
5. Janik, A., Qiu, X., Lane, R., Popova, V., Drevets, W. C., Canuso, C. M., Macaluso, M., Mattingly, G. W., Shelton, R. C., Zajecka, J. M., & Fu, D.-J. (2025). Esketamine Monotherapy in Adults With Treatment-Resistant Depression: A Randomized Clinical Trial. JAMA Psychiatry, 82(9), 877–887. https://doi.org/10.1001/jamapsychiatry.2025.1317
6. Sanacora, G., Ahmed, M., Brown, B., Cabrera, P., Doherty, T., Himedan, M., Kern, D. M., Lim, L., Lopena, O., Ronaldo R. Naranjo, J., Nuamah, I., Sarayani, A., Turkoz, I., & Bowrey, H. E. (2025). Real-World Safety of Esketamine Nasal Spray: A Comprehensive Analysis Almost 5 Years After First Approval. American Journal of Psychiatry. https://doi.org/10.1176/appi.ajp.20240655
7. Naudet, F., Pellen, C., Fodor, L. A., Gastaldon, C., Barbui, C., Turner, E. H., Le Pabic, E., & Cristea, I. A. (2025). Efficacy and safety of esketamine for « treatment resistant depression »: Registered report for a systematic review with an individual patient data meta-analysis of randomized, double-blind, placebo-controlled trials. BMC Medicine, 23(1), 677. https://doi.org/10.1186/s12916-025-04435-x
8. Vasiliu, O. (2023). Esketamine for treatment-resistant depression: A review of clinical evidence (Review). Experimental and Therapeutic Medicine, 25(3), 111. https://doi.org/10.3892/etm.2023.11810
9. Papakostas, G. I., Salloum, N. C., Hock, R. S., Jha, M. K., Murrough, J. W., Mathew, S. J., Iosifescu, D. V., & Fava, M. (2020). Efficacy of Esketamine Augmentation in Major Depressive Disorder: A Meta-Analysis. The Journal of Clinical Psychiatry, 81(4), 19r12889. https://doi.org/10.4088/JCP.19r12889
10. Floriano, I., Silvinato, A., & Bernardo, W. M. (2023). The use of esketamine in the treatment of patients with severe depression and suicidal ideation: Systematic review and meta-analysis. Revista Da Associação Médica Brasileira, 69(4), e2023D694. https://doi.org/10.1590/1806-9282.2023d694
11. McIntyre, R. S., Rosenblat, J. D., Nemeroff, C. B., Sanacora, G., Murrough, J. W., Berk, M., Brietzke, E., Dodd, S., Gorwood, P., Ho, R., Iosifescu, D. V., Lopez Jaramillo, C., Kasper, S., Kratiuk, K., Lee, J. G., Lee, Y., Lui, L. M. W., Mansur, R. B., Papakostas, G. I., … Stahl, S. (2021). Synthesizing the Evidence for Ketamine and Esketamine in Treatment-Resistant Depression: An International Expert Opinion on the Available Evidence and Implementation. The American Journal of Psychiatry, 178(5), 383–399. https://doi.org/10.1176/appi.ajp.2020.20081251
12. Cristea, I. A., Plöderl, M., & Naudet, F. (2025). Esketamine for Treatment-Resistant Depression. JAMA Psychiatry. https://doi.org/10.1001/jamapsychiatry.2025.3242
13. FDA approval of CAPLYTAreg (lumateperone) has the potential to reset treatment expectations, offering hope for remission in adults with major depressive disorder. (2025, November 6). JNJ.com. https://www.jnj.com/media-center/press-releases/fda-approval-of-caplyta-lumateperone-has-the-potential-to-reset-treatment-expectations-offering-hope-for-remission-in-adults-with-major-depressive-disorder
14. US Food and Drug Administration (2025). CAPLYTAreg (lumateperone) capsules, for oral use. https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/209500s016lbl.pdf
15. IsHak, W. W., Hirsch, D., Renteria, S., Totlani, J., Murphy, N., Chang, T., Abdelsalam, R., Salem, M., Meyer, A., Keerthana, S., Liu, A., Contreras, L., Tadros, E., Hedrick, R., Danovitch, I., & Pechnick, R. N. (2025). Depressive disorders: Systematic review of approved psychiatric medications (2009-April 2025) and pipeline phase 3 medications. BMC Psychiatry, 25, 939. https://doi.org/10.1186/s12888-025-07141-3
16. Jha, M. K., & Mathew, S. J. (2023). Pharmacotherapies for Treatment-Resistant Depression: How Antipsychotics Fit in the Rapidly Evolving Therapeutic Landscape. American Journal of Psychiatry. https://doi.org/10.1176/appi.ajp.20230025
17. Thase, M. (n.d.). Adjunctive Antipsychotic Therapies in the Treatment of Major Depressive Disorder. Psychiatry Advisor. Retrieved December 8, 2025, from https://www.psychiatryadvisor.com/cch/major-depressive-disorder-second-generation-antipsychotics-brexpiprazole/
18. Durgam, S., Earley, W., Kozauer, S. G., Chen, C., Lakkis, H., Edwards, J. B., & Stahl, S. (2024). Lumateperone as adjunctive therapy in patients with major depressive disorder: Results from a randomized, double-blind, phase 3 trial. Neuroscience Applied, 3, 104586. https://doi.org/10.1016/j.nsa.2024.104586
19. Durgam, S., Earley, W. R., Kozauer, S. G., Chen, C., Edwards, J. B., Jain, R., & Correll, C. U. (n.d.). Adjunctive Lumateperone 42 mg in the Treatment of Major Depressive Disorder: A Pooled Analysis of 2 Phase 3 Randomized Controlled Trials.
20. Neuronetics. (2024, March 25). NeuroStarreg Advanced Therapy Receives FDA Clearance as a First-Line Add-On Treatment for Adolescents with Depression. GlobeNewswire News Room. https://www.globenewswire.com/en/news-release/2024/03/25/2851611/0/en/NeuroStar-Advanced-Therapy-Receives-FDA-Clearance-as-a-First-Line-Add-On-Treatment-for-Adolescents-with-Depression.html
21. Mandeville, R. (2025, August 25). FDA clearance of TMS Therapy for Adolescents. MagVenture US. https://magventure.com/us/fda-clearance-to-expand-tms-therapy-indication-for-adolescents-aged-15-21/
22. AG, neurocare group. (2025, September 10). Neurocare group’s Apollo TMS Therapy Devices Now Have FDA Clearance to Treat Adolescents Suffering With Major Depression. GlobeNewswire News Room. https://www.globenewswire.com/news-release/2025/09/10/3147821/0/en/neurocare-group-s-Apollo-TMS-Therapy-Devices-Now-Have-FDA-Clearance-to-Treat-Adolescents-Suffering-With-Major-Depression.html
23. BrainsWay Receives FDA Clearance of Deep TMSTM as Adjunct Therapy for Major Depressive Disorder (MDD) in Adolescents Aged 15 to 21 – BrainsWay. (n.d.). Retrieved December 8, 2025, from https://investors.brainsway.com/news-releases/news-release-details/brainsway-receives-fda-clearance-deep-tmstm-adjunct-therapy/
24. Hanlon, C. A., Roth, Y., Bermudes, R. A., Brink, E., Davis, M., DeBrocco, D., Ellis, S., Jones, L., Khan, A., MacMillan, C., Mudunuru, A. K., Muir, O., Pell, G. S., Prestley, T., Reddy, M. S., Sisko, E., Seplow, S., Slomowitz, N., Stein, A., … Tendler, A. (2025, October 22). Accelerated Deep TMS for Depression: Results from a Multisite, Randomized Non-Inferiority Trial (SSRN Scholarly Paper 5583382). https://doi.org/10.2139/ssrn.5583382 (Pre-published)
25. FDA Approves World’s First At-Home Brain Stimulation Treatment for Depression. (2025, December 11). https://www.flowneuroscience.com/fda-approved-lp-2/
26. Woodham, R. D., Selvaraj, S., Lajmi, N., Hobday, H., Sheehan, G., Ghazi-Noori, A.-R., Lagerberg, P. J., Rizvi, M., Kwon, S. S., Orhii, P., Maislin, D., Hernandez, L., Machado-Vieira, R., Soares, J. C., Young, A. H., & Fu, C. H. Y. (2025). Home-based transcranial direct current stimulation treatment for major depressive disorder: A fully remote phase 2 randomized sham-controlled trial. Nature Medicine, 31(1), 87–95. https://doi.org/10.1038/s41591-024-03305-y
27. Woodham, R., Rimmer, R. M., Mutz, J., & Fu, C. H. Y. (2021). Is tDCS a potential first line treatment for major depression? International Review of Psychiatry (Abingdon, England), 33(3), 250–265. https://doi.org/10.1080/09540261.2021.1879030
28. Ironside, M., Browning, M., Ansari, T. L., Harvey, C. J., Sekyi-Djan, M. N., Bishop, S. J., Harmer, C. J., & O’Shea, J. (2019). Effect of Prefrontal Cortex Stimulation on Regulation of Amygdala Response to Threat in Individuals With Trait Anxiety: A Randomized Clinical Trial. JAMA Psychiatry, 76(1), 71–78. https://doi.org/10.1001/jamapsychiatry.2018.2172
29. Keeser, D., Meindl, T., Bor, J., Palm, U., Pogarell, O., Mulert, C., Brunelin, J., Möller, H.-J., Reiser, M., & Padberg, F. (2011). Prefrontal transcranial direct current stimulation changes connectivity of resting-state networks during fMRI. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 31(43), 15284–15293. https://doi.org/10.1523/JNEUROSCI.0542-11.2011
30. FDA Approves Expanded Indication for UZEDYreg (risperidone) Extended-Release Injectable Suspension as a Treatment for Adults Living with Bipolar I Disorder. (n.d.). Retrieved December 8, 2025, from https://ir.tevapharm.com/news-and-events/press-releases/press-release-details/2025/FDA-Approves-Expanded-Indication-for-UZEDY-risperidone-Extended-Release-Injectable-Suspension-as-a-Treatment-for-Adults-Living-with-Bipolar-I-Disorder/default.aspx
31. US Food and Drug Administration (2025). UZEDY (risperidone) extended-release injectable suspension, for subcutaneous use. https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/213586s008s009lbl.pdf
32. US Food and Drug Administration (2025). UZEDYreg (risperidone) extended-release injectable suspension. https://www.uzedy.com/globalassets/uzedy/prescribing-information.pdf#xd_co_f=MGJhYmRmNDEtNmU1Ni00ZjdhLTk4MWQtOWZiNzcxNjA0NzA0~
33. US Food and Drug Administration (Wed, 08/27/2025 – 14:52). FDA removes risk evaluation and mitigation strategy (REMS) program for the antipsychotic drug Clozapine. FDA. https://www.fda.gov/drugs/drug-safety-and-availability/fda-removes-risk-evaluation-and-mitigation-strategy-rems-program-antipsychotic-drug-clozapine
34. US Food and Drug Administration (Tue, 02/25/2025 – 09:29, Tue, 02/25/2025 – 09:29). Frequently Asked Questions | Clozapine REMS Modification. Clozapine REMS Modification; FDA. https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/frequently-asked-questions-clozapine-rems-modification
35. CLOZARILreg (clozapine) tablets, for oral use. (n.d.). Retrieved December 8, 2025, from https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/019758s106lbl.pdf
36. US Food and Drug Administration (Tue, 07/01/2025 – 11:32). FDA requires expanded labeling about weight loss risk in patients younger than 6 years taking extended-release stimulants for ADHD. FDA. https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-expanded-labeling-about-weight-loss-risk-patients-younger-6-years-taking-extended
37. Company, E. L. and. (2025, July 9). FDA approves updated label for Lilly’s Kisunla (donanemab-azbt) with new dosing in early symptomatic Alzheimer’s disease. https://investor.lilly.com/news-releases/news-release-details/fda-approves-updated-label-lillys-kisunla-donanemab-azbt-new
38. US Food and Drug Administration (2025). KISUNLA (donanemab-azbt) injection, for intravenous use. https://www.accessdata.fda.gov/drugsatfda_docs/label/2025/761248s004lbl.pdf
39. Rabinovici, G. D., Selkoe, D. J., Schindler, S. E., Aisen, P., Apostolova, L. G., Atri, A., Greenberg, S. M., Hendrix, S. B., Petersen, R. C., Weiner, M., Salloway, S., & Cummings, J. (2025). Donanemab: Appropriate use recommendations. The Journal of Prevention of Alzheimer’s Disease, 12(5), 100150. https://doi.org/10.1016/j.tjpad.2025.100150
40. Sims, J. R., Zimmer, J. A., Evans, C. D., Lu, M., Ardayfio, P., Sparks, J., Wessels, A. M., Shcherbinin, S., Wang, H., Monkul Nery, E. S., Collins, E. C., Solomon, P., Salloway, S., Apostolova, L. G., Hansson, O., Ritchie, C., Brooks, D. A., Mintun, M., Skovronsky, D. M., & TRAILBLAZER-ALZ 2 Investigators. (2023). Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA, 330(6), 512–527. https://doi.org/10.1001/jama.2023.13239
41. Ross, E. L., Weinberg, M. S., & Arnold, S. E. (2022). Cost-effectiveness of Aducanumab and Donanemab for Early Alzheimer Disease in the US. JAMA Neurology, 79(5), 478–487. https://doi.org/10.1001/jamaneurol.2022.0315
42. Boustani, M., Doty, E. G., Garrison, L. P., Smolen, L. J., Belger, M., Klein, T. M., Murphy, D. R., Burge, R., Wall, J. K., & Johnston, J. A. (2022). Assessing the Cost-effectiveness of a Hypothetical Disease-modifying Therapy With Limited Duration for the Treatment of Early Symptomatic Alzheimer Disease. Clinical Therapeutics, 44(11), 1449–1462. https://doi.org/10.1016/j.clinthera.2022.09.008
43. Mattke, S., Ozawa, T., & Hanson, M. (2024). Implications of treatment duration and frequency for value and cost-effective price of Alzheimer treatments. Journal of Managed Care & Specialty Pharmacy, 30(10), 1087–1094. https://doi.org/10.18553/jmcp.2024.24116
44. Office of the Commissioner. US Food and Drug Administration (Wed, 05/28/2025 – 11:33, Wed, 05/28/2025 – 11:33). FDA Clears First Blood Test Used in Diagnosing Alzheimer’s Disease. FDA Clears First Blood Test Used in Diagnosing Alzheimer’s Disease; FDA. https://www.fda.gov/news-events/press-announcements/fda-clears-first-blood-test-used-diagnosing-alzheimers-disease
45. Tonix Pharmaceuticals (2025). Tonix Pharmaceuticals Announces FDA Approval of TonmyaTM (cyclobenzaprine HCl sublingual tablets) for the Treatment of Fibromyalgia. https://ir.tonixpharma.com/news-events/press-releases/detail/1585/tonix-pharmaceuticals-announces-fda-approval-of
46. Lederman, S., Arnold, L. M., Vaughn, B., Engels, J. M., Kelley, M., & Sullivan, G. M. (2025). Pain relief by targeting nonrestorative sleep in fibromyalgia: A phase 3 randomized trial of bedtime sublingual cyclobenzaprine. Pain Medicine, pnaf089. https://doi.org/10.1093/pm/pnaf089
47. Perrot, S., & Russell, I. j. (2014). More ubiquitous effects from non-pharmacologic than from pharmacologic treatments for fibromyalgia syndrome: A meta-analysis examining six core symptoms. European Journal of Pain, 18(8), 1067–1080. https://doi.org/10.1002/ejp.564
48. Häuser, W., Petzke, F., & Sommer, C. (2010). Comparative Efficacy and Harms of Duloxetine, Milnacipran, and Pregabalin in Fibromyalgia Syndrome. The Journal of Pain, 11(6), 505–521. https://doi.org/10.1016/j.jpain.2010.01.002
49. Farag, H. M., Yunusa, I., Goswami, H., Sultan, I., Doucette, J. A., & Eguale, T. (2022). Comparison of Amitriptyline and US Food and Drug Administration–Approved Treatments for Fibromyalgia. JAMA Network Open, 5(5), e2212939. https://doi.org/10.1001/jamanetworkopen.2022.12939
50. Lederman, S., Arnold, L. M., Vaughn, B., Kelley, M., & Sullivan, G. M. (2023). Efficacy and Safety of Sublingual Cyclobenzaprine for the Treatment of Fibromyalgia: Results From a Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Care & Research, 75(11), 2359–2368. https://doi.org/10.1002/acr.25142
51. Kaufman, M. B., PharmD, & BCGP. (n.d.). A New Treatment for Fibromyalgia? – Page 3 of 4. The Rheumatologist. Retrieved December 9, 2025, from https://www.the-rheumatologist.org/article/a-new-treatment-for-fibromyalgia/
52. Indivior. (2025, February 24). Indivior Announces FDA Approval of Label Changes for SUBLOCADEreg (buprenorphine extended-release) Injection. https://www.indivior.com/en/media/press-releases/indivior-announces-fda-approval-of-label-changes-for-sublocade-injection
53. US Food and Drug Administration (2023). SUBLOCADE (buprenorphine extended-release) injection, for subcutaneous use, CIII. https://www.accessdata.fda.gov/drugsatfda_docs/label/2023/209819s028lbl.pdf
